Welche Nebenwirkungen haben Chemotherapien?
Bei der Chemotherapie muss man akute und langfristige Nebenwirkungen unterscheiden. Kurzfristige Nebenwirkungen sind vor allem die Störung der Blutbildung, die Schädigung der peripheren Nerven und Haarverlust; Übelkeit und Erbrechen spielen heute bei den zur Behandlung des DLBCL eingesetzten Medikamenten dank wirksamer Gegenmittel gegen diese Nebenwirkung (= Antiemetika) keine Rolle mehr. Langfristige Nebenwirkungen betreffen die Funktion von Herz und Keimdrüsen und ein erhöhtes Risiko, an weiteren bösartigen
Tumoren zu erkranken.
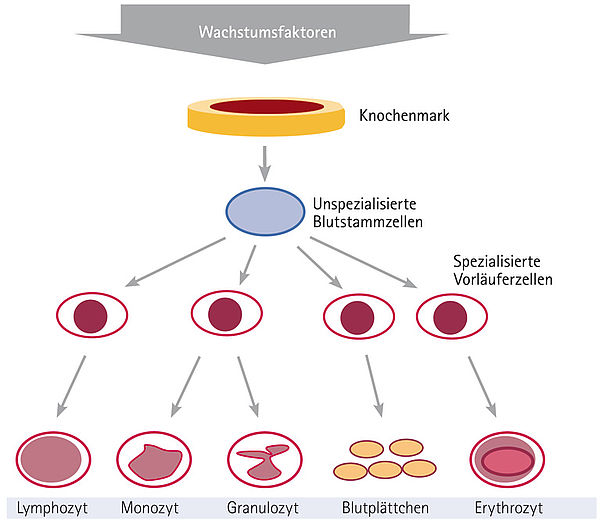
Die Standard-Immunchemotherapie mit CHOP und Rituximab wird im Allgemeinen bis ins hohe Alter gut vertragen. Durch die Wirkstoffe der CHOP-Chemotherapie kommt es akut zur Unterdrückung des Knochenmarks mit verminderter Bildung von weißen und roten Blutkörperchen sowie Blutplättchen (= Myelosuppression). Die Myelosuppression beginnt meistens 6-8 Tage
nach Beginn der Chemotherapie, erreicht ihre stärkste Ausprägung am Tag 10, danach kommt es zur raschen Erholung. Deshalb sollte das Blutbild nach jedem Chemotherapie-Zyklus ein bis zwei Mal pro Woche kontrolliert werden.
Ein erneuter Zyklus darf erst begonnen werden, wenn sich das Knochenmark weitgehend von der letzten Therapie erholt hat. Ob die Dosis einzelner Wirkstoffe reduziert werden muss, hängt davon ab, wie stark das Knochenmark geschädigt war und wie lange diese Schädigung angehalten hat.
Während der Immunchemotherapie ist das Risiko einer Infektion abhängig von der Zahl der weißen Blutkörperchen deutlich erhöht. Daher sollten ältere Menschen (über 60 Jahre) nach R-CHOP und jüngere Menschen nach R-CHOP-14 oder R-CHOEP-14 Wachstumsfaktoren (G-CSF = Granulozyten-Kolonien-stimulierender Faktor) erhalten. G-CSF wird als Spritze (= Injektion) verabreicht und bewirkt, dass die weißen Blutkörperchen nicht so stark abfallen und schneller wieder ansteigen. Zusätzlich wird eine Vorbeugung vor Infekten empfohlen. Sie sollte bis zu vier Wochen nach Abschluss der Immunchemotherapie gegeben werden und aus den Wirkstoffen Aciclovir gegen Herpes- und CMV-Infektionen sowie Cotrimoxazol gegen Lungeninfektionen bestehen. Fallen die weißen Blutkörperchen unter 1.000 oder die Granulozyten unter 500 pro Kubikmillimeter (mm3), so sollte zusätzlich ein Antibiotikum (z.B. Ciprofloxazin)
bis zur Erholung der weißen Blutkörperchen (meistens am Tag 12 nach CHOP) gegeben werden. Die verminderte Produktion roter Blutkörperchen unter Chemotherapie führt nur selten zu einer Blutarmut (= Anämie), die eine Transfusion roter Blutkörperchen nötig macht. Verminderungen der Blutplättchen (= Thrombozyten) in einem Ausmaß, dass Thrombozyten übertragen
werden müssen, sind extrem selten.
Eine Schädigung der peripheren Nerven, die sich durch Kribbeln oder Taubheitsgefühle in Händen und Füßen (= Polyneuropathie) bemerkbar macht, wird durch den Wirkstoff Vincristin hervorgerufen und betrifft vor allem ältere Patientinnen und Patienten. Da diese Schädigungen mit jedem weiteren CHOP-Zyklus rasant zunehmen können, muss das Behandlungsteam schon
beim Auftreten erster Beschwerden informiert werden. Gegebenenfalls wird dann beim nächsten Chemotherapiezyklus die Vincristin-Dosis reduziert oder Vincristin ganz abgesetzt.
Neben den Blutbildveränderungen und der damit erhöhten Infektionsgefahr stellt der durch die Chemotherapie bedingte Haarausfall (= Alopezie) für viele Patientinnen und Patienten die zweitgrößte Belastung dar. Allerdings fangen die Haare bereits wenige Wochen nach dem Ende der Chemotherapie wieder an zu wachsen.
Langfristige Nebenwirkungen können die Funktion der Keimdrüsen (= Hoden, Eierstöcke), des Herzens und des Knochenmarks betreffen. Bei Patienten mit Kinderwunsch sollte daher über die Gewinnung und Lagerung von Spermien bzw. Eierstockgewebe (= Asservierung) und/oder protektive Maßnahmen für die Eierstöcke gesprochen werden. Insbesondere Doxorubicin (das
„H“ im CHOP-Schema) kann zu einer Schädigung der Herzmuskelkraft führen, was vor allem bei bereits vorgeschädigten Herzen beobachtet wird. Deshalb sollte die Herzfunktion (Herzultraschall, EKG) bereits vor Beginn der Therapie als auch in regelmäßigen Abständen danach überprüft werden.